
Decoding the complexity of Kidney Disease
March is National Kidney Month, a time to recognize a growing global crisis. Today, kidney disease affects over 850 million people and is projected to become the 5th leading cause of death worldwide. Every year, 11 million lives are lost to a disease whose critical signals often remain hidden within the structural complexity of our cells.
At OmnibusX, we provide tools that support kidney research, from open access to curated public datasets to interactive analysis tools and multi-omics workflows spanning bulk RNA-seq, single-cell kidney atlases, and spatial niches.

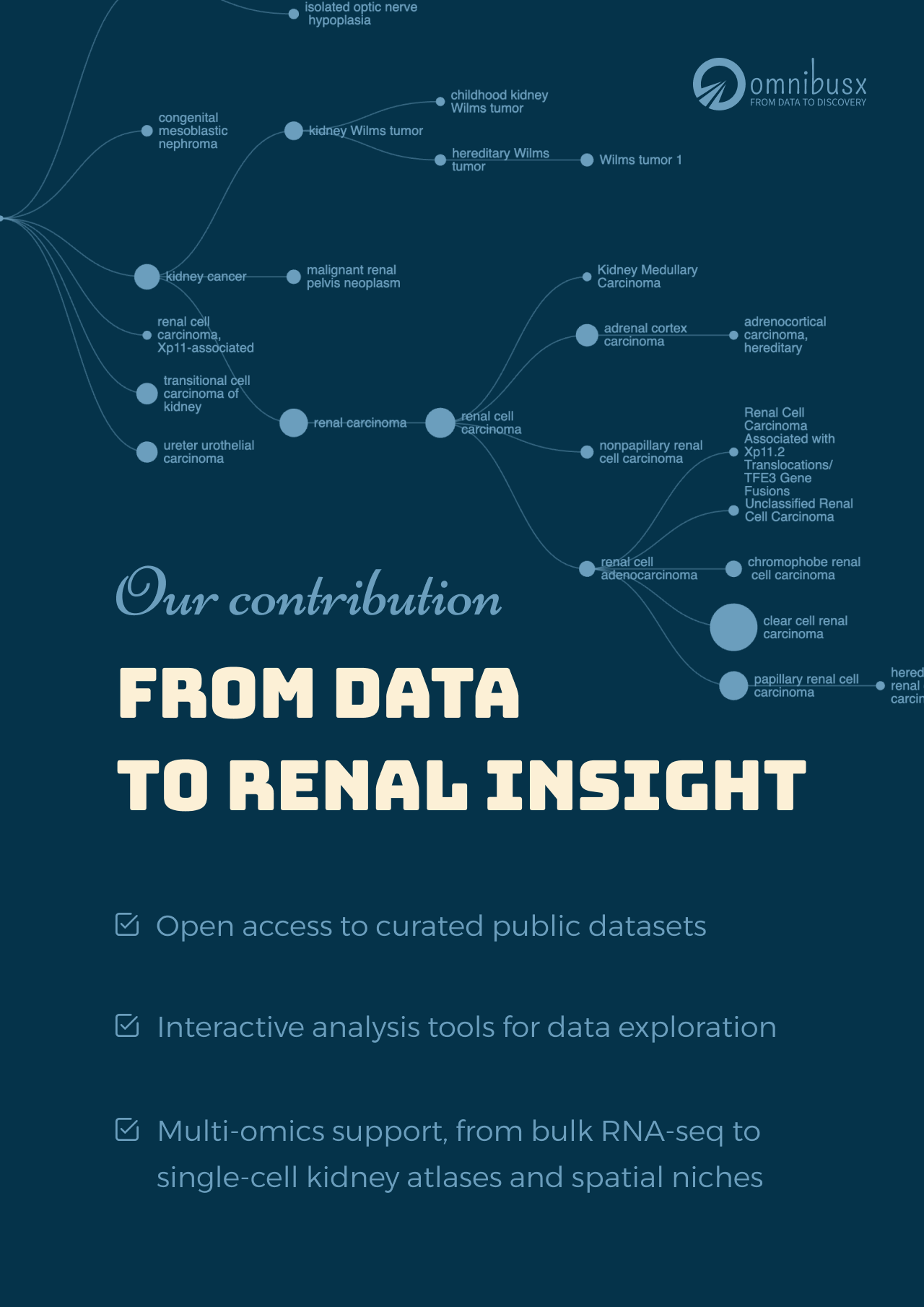
To mark this month, we are sharing a selection of curated public kidney datasets that you can explore in real time, with no coding and no data processing required:
1. Analysis of individual patient pathway coordination in a cross-species single-cell kidney atlas

Abstract
The use of single-cell RNA sequencing in clinical and translational research is limited by the challenge of identifying cell-type-specific, targetable molecular changes in individual patients and cross-species differences. Here we created an integrated single-cell kidney atlas including over 1 million cells from 140 samples, defining more than 70 conserved cell states in human and rodent models. We developed CellSpectra, a computational tool that quantifies changes in gene expression coordination across cellular functions, which we applied to kidney and lung cancer data. This tool powers our patient-level single-cell functional profiling report, which highlights cell-type-specific changes in the coordination of pathway gene expression in individuals. Our cross-species atlas facilitates the selection of a rodent model that closely reflects the cellular and pathway-level signatures observed in patient samples, advancing the application of single-cell methodologies in clinical precision medicine. Finally, using experimental models, we demonstrate how our informatics approach can be applied for the potential selection of suitable therapeutics.

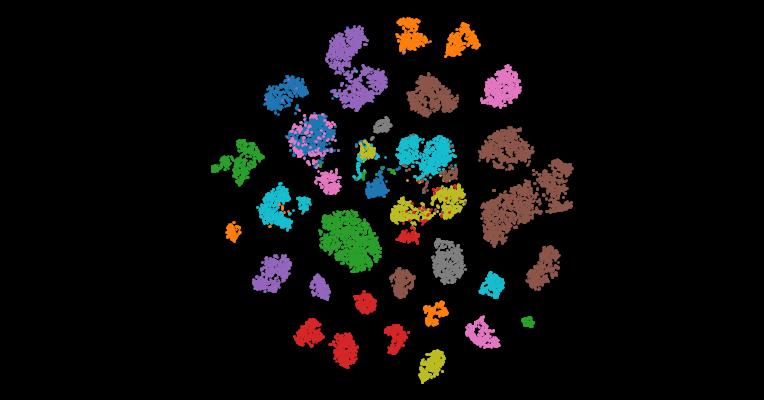
2. An atlas of healthy and injured cell states and niches in the human kidney
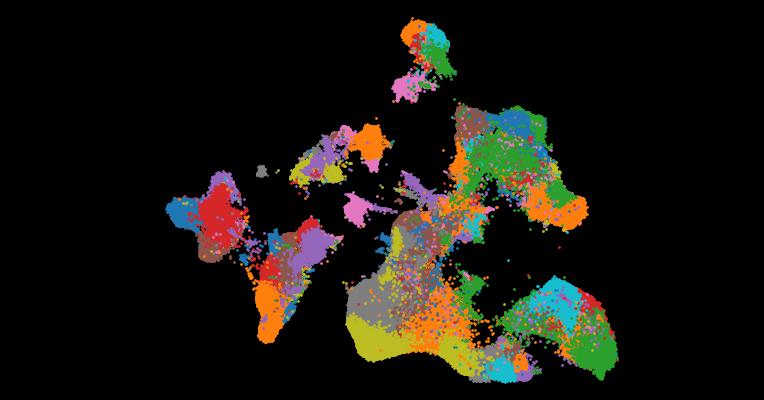
Abstract
Understanding kidney disease relies on defining the complexity of cell types and states, their associated molecular profiles and interactions within tissue neighbourhoods1. Here we applied multiple single-cell and single-nucleus assays (>400,000 nuclei or cells) and spatial imaging technologies to a broad spectrum of healthy reference kidneys (45 donors) and diseased kidneys (48 patients). This has provided a high-resolution cellular atlas of 51 main cell types, which include rare and previously undescribed cell populations. The multi-omic approach provides detailed transcriptomic profiles, regulatory factors and spatial localizations spanning the entire kidney. We also define 28 cellular states across nephron segments and interstitium that were altered in kidney injury, encompassing cycling, adaptive (successful or maladaptive repair), transitioning and degenerative states. Molecular signatures permitted the localization of these states within injury neighbourhoods using spatial transcriptomics, while large-scale 3D imaging analysis (around 1.2 million neighbourhoods) provided corresponding linkages to active immune responses. These analyses defined biological pathways that are relevant to injury time-course and niches, including signatures underlying epithelial repair that predicted maladaptive states associated with a decline in kidney function. This integrated multimodal spatial cell atlas of healthy and diseased human kidneys represents a comprehensive benchmark of cellular states, neighbourhoods, outcome-associated signatures and publicly available interactive visualizations.
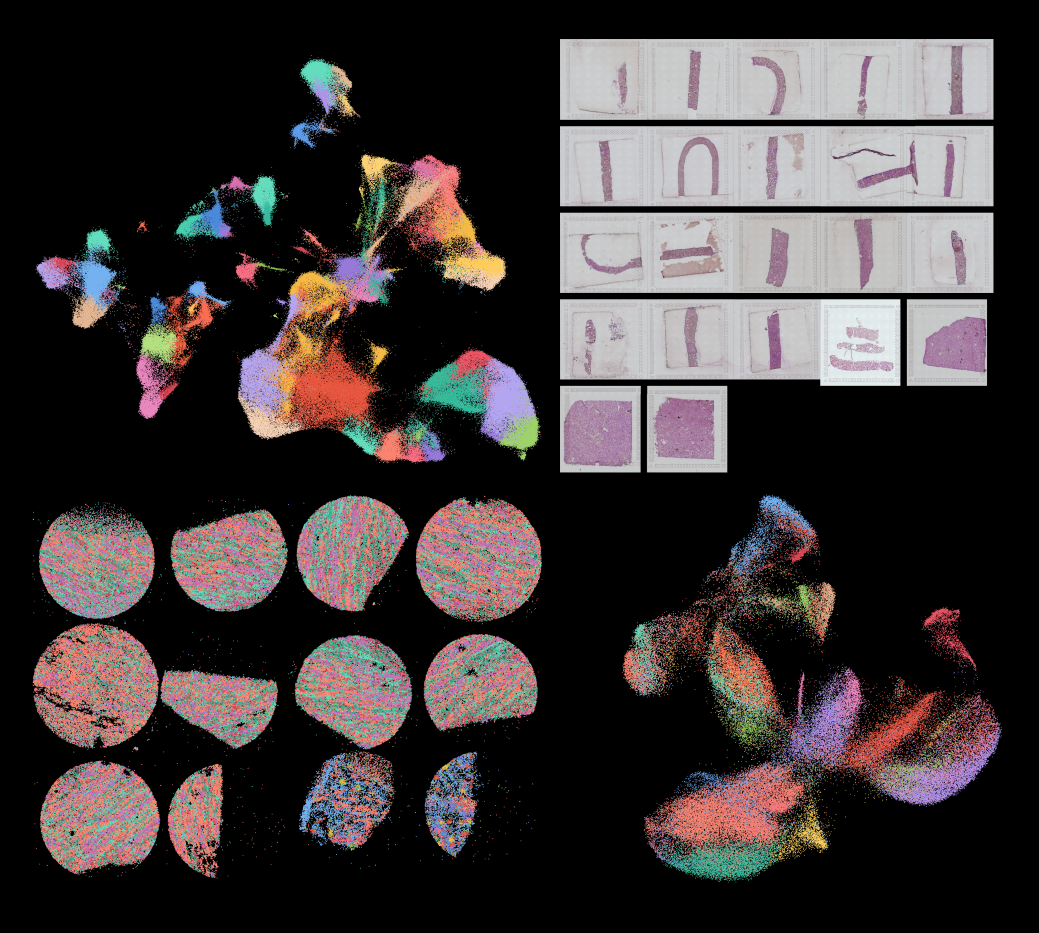
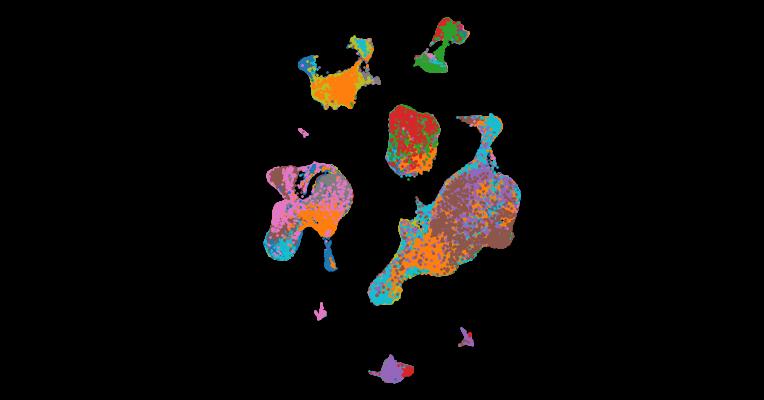
3. Automatic cell-type harmonization and integration across Human Cell Atlas datasets

Abstract
Harmonizing cell types across the single-cell community and assembling them into a common framework is central to building a standardized Human Cell Atlas. Here, we present CellHint, a predictive clustering tree-based tool to resolve cell-type differences in annotation resolution and technical biases across datasets. CellHint accurately quantifies cell-cell transcriptomic similarities and places cell types into a relationship graph that hierarchically defines shared and unique cell subtypes. Application to multiple immune datasets recapitulates expert-curated annotations. CellHint also reveals underexplored relationships between healthy and diseased lung cell states in eight diseases. Furthermore, we present a workflow for fast cross-dataset integration guided by harmonized cell types and cell hierarchy, which uncovers underappreciated cell types in adult human hippocampus. Finally, we apply CellHint to 12 tissues from 38 datasets, providing a deeply curated cross-tissue database with ∼3.7 million cells and various machine learning models for automatic cell annotation across human tissues.

4. Mapping single-cell transcriptomes in the intra- tumoral and associated territories of kidney cancer
Abstract
Tumor behavior is intricately dependent on the oncogenic properties of cancer cells and their multi-cellular interactions. To understand these dependencies within the wider microenvironment, we studied over 270,000 single-cell transcriptomes and 100 microdissected whole exomes from 12 patients with kidney tumors, prior to validation using spatial transcriptomics. Tissues were sampled from multiple regions of the tumor core, the tumor-normal interface, normal surrounding tissues, and peripheral blood. We find that the tissue-type location of CD8+ T cell clonotypes largely defines their exhaustion state with intra-tumoral spatial heterogeneity that is not well explained by somatic heterogeneity. De novo mutation calling from single-cell RNA-sequencing data allows us to broadly infer the clonality of stromal cells and lineage-trace myeloid cell development. We report six conserved meta-programs that distinguish tumor cell function, and find an epithelial-mesenchymal transition meta-program highly enriched at the tumor-normal interface that co-localizes with IL1B-expressing macrophages, offering a potential therapeutic target.
5. Spatially resolved human kidney multi-omics single cell atlas highlights the key role of the fibrotic microenvironment in kidney disease progression

Abstract
Kidneys possess one of the most intricate three-dimensional cellular structures in the body, yet the spatial and molecular principles of kidney health and disease remain inadequately understood. Here, we have generated high-quality datasets for 81 samples, including single cell (sc), single nuclear (sn), spot level (Visium) and single cell resolution (CosMx) spatial (sp)-RNA expression, and sn open chromatin, capturing cells from healthy, diabetic, and hypertensive diseased human kidneys. By combining the snRNA, snATAC and scRNA sequencing we identify cell types and map these cell types to their locations within the tissue. Unbiased deconvolution of the spatial data identifies 4 distinct spatial microenvironments: glomerular, immune, tubule and fibrotic. We describe the complex, heterogenous cellular and spatial organization of human microenvironments in health and disease. Further, we find that the fibrotic microenvironment spatial gene signature is not only able to molecularly classify human kidneys, but it also offers an improved prognosis prediction compared to traditional histopathological analysis. We provide a comprehensive spatially resolved molecular roadmap of the human kidney and the fibrotic process, demonstrating the clinical utility of spatial transcriptomics.

6. Single cell derived mRNA signals across human kidney tumors

Abstract
Tumor cells may share some patterns of gene expression with their cell of origin, providing clues into the differentiation state and origin of cancer. Here, we study the differentiation state and cellular origin of 1300 childhood and adult kidney tumors. Using single cell mRNA reference maps of normal tissues, we quantify reference “cellular signals” in each tumor. Quantifying global differentiation, we find that childhood tumors exhibit fetal cellular signals, replacing the presumption of “fetalness” with a quantitative measure of immaturity. By contrast, in adult cancers our assessment refutes the suggestion of dedifferentiation towards a fetal state in most cases. We find an intimate connection between developmental mesenchymal populations and childhood renal tumors. We demonstrate the diagnostic potential of our approach with a case study of a cryptic renal tumor. Our findings provide a cellular definition of human renal tumors through an approach that is broadly applicable to human cancer.
7. The Cancer Genome Atlas Pan-Cancer analysis project
Abstract
The Cancer Genome Atlas (TCGA) Research Network has profiled and analyzed large numbers of human tumors to discover molecular aberrations at the DNA, RNA, protein and epigenetic levels. The resulting rich data provide a major opportunity to develop an integrated picture of commonalities, differences and emergent themes across tumor lineages. The Pan-Cancer initiative compares the first 12 tumor types profiled by TCGA. Analysis of the molecular aberrations and their functional roles across tumor types will teach us how to extend therapies effective in one cancer type to others with a similar genomic profile.
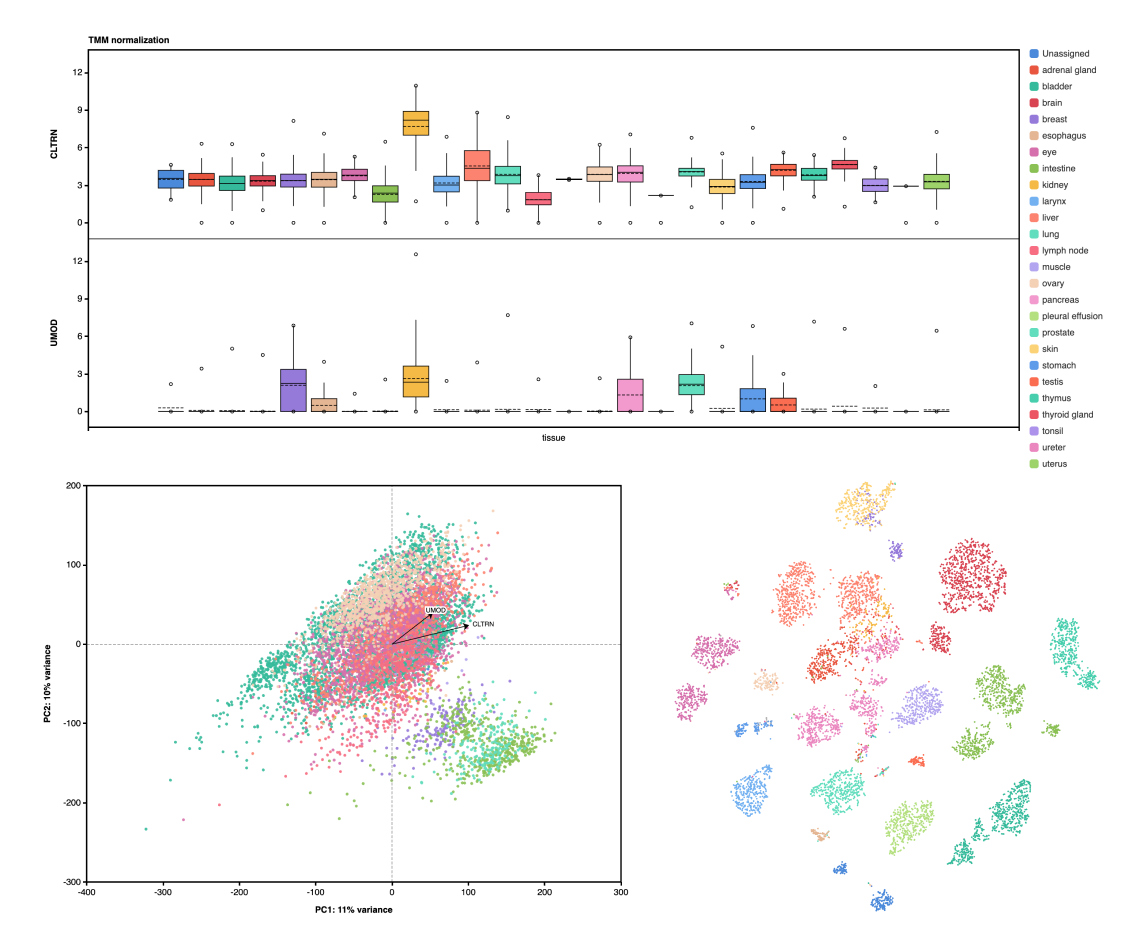
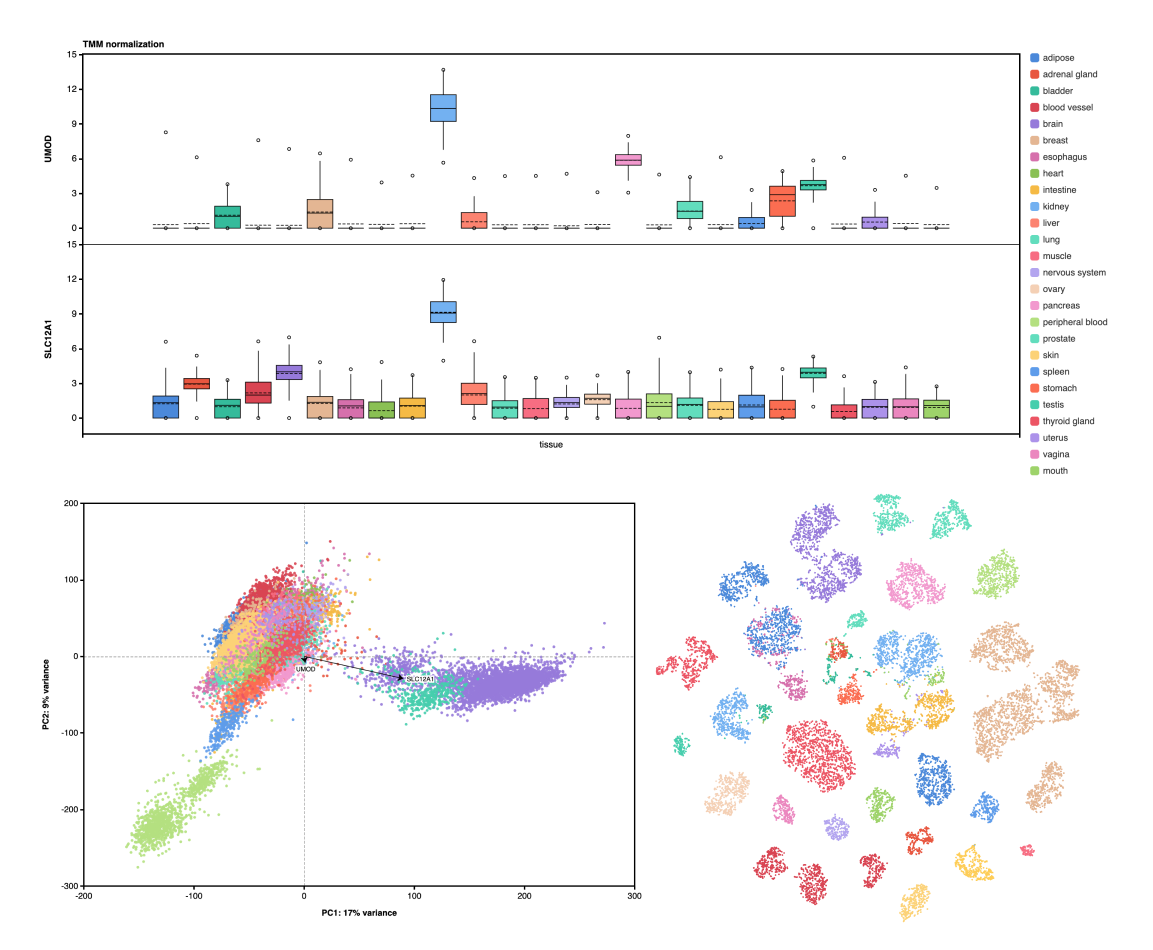
8. The GTEx Consortium atlas of genetic regulatory effects across human tissues
Abstract
The Genotype-Tissue Expression (GTEx) project was established to characterize genetic effects on the transcriptome across human tissues and to link these regulatory mechanisms to trait and disease associations. Here, we present analyses of the version 8 data, examining 15,201 RNA-sequencing samples from 49 tissues of 838 postmortem donors. We comprehensively characterize genetic associations for gene expression and splicing in cis and trans, showing that regulatory associations are found for almost all genes, and describe the underlying molecular mechanisms and their contribution to allelic heterogeneity and pleiotropy of complex traits. Leveraging the large diversity of tissues, we provide insights into the tissue specificity of genetic effects and show that cell type composition is a key factor in understanding gene regulatory mechanisms in human tissues.
You can also analyze your own kidney data with our multi-omics application here: https://omnibusx.com/apps